

Introduction
EU good manufacturing observe (GMP) Annex 11 regulation on computerized methods has been efficient since 1992.1 Following the info falsification uncovered at Ready Laboratories in 2005,2 many information integrity (DI) points involving computerized methods have been uncovered throughout inspections.
In 2008, a proposed replace to Annex 11 to bolster DI controls was issued for trade remark.3 The revised model was issued in 20114 together with an up to date EU GMP Chapter 4 on documentation.5 EU Annex 11 and Chapter 4 are additionally a part of Pharmaceutical Inspection Conference Scheme (PIC/S) GMP, which might be relevant in international locations comparable to Japan, South Korea, Australia, New Zealand, and many others.
In 2022, an idea paper on the revision of Annex 11 was issued by the European Medicines Company (EMA) and PIC/S for trade remark,6 and in July 2025, the revised variations of Annex 11 and Chapter 4 have been issued for stakeholder feedback.7 On the identical time, a brand new Annex 22 on synthetic intelligence (AI) was additionally issued for stakeholder remark.7 We is not going to be discussing Annex 22 right here.
This text gives detailed analyses of Annex 11 revision and the relevant clauses of Chapter 4 that influence computerized methods. The detailed dialogue on this article consists of:
One of many main issues with the revision is that it isn’t straightforward to map to the present model of the regulation. Sections seem to have been chosen by pulling names out of a hat.
Why talk about Chapter 4 on documentation?
As famous above, Annex 11 and Chapter 4 are revised in parallel. The rationale is:
To Totally Perceive Annex 11, You Should Perceive Chapter 4.
What’s the rationale for needing to grasp the implications of Chapter 4 on Annex 11? The clue is within the chapter title: Documentation. This consists of danger administration, digital information, plus the underlying information/metadata, DI, information governance, ALCOA++ standards, grasp paperwork, hybrid methods, signatures on GMP paperwork and file retention.
, Chapter 4 (documentation), Annex 15 (qualification and validation) and Chapter 7 (outsourced activities). Credit: Bob McDowall.")
Determine 1: Relationships between Annex 11 (computerized methods), Chapter 4 (documentation), Annex 15 (qualification and validation) and Chapter 7 (outsourced actions). Credit score: Bob McDowall.
Contemplating the 2 laws individually signifies that you miss the large image proven in Determine 1. Due to this fact, this important evaluation of the revised Annex 11 will embrace acceptable sections of Chapter 4 as computerized methods create, interpret, report and retailer regulatory paperwork, information and information. Sections of each paperwork not mentioned are proven in purple in Determine 1. Readers should additionally bear in mind that there are necessities in Annex 15 on qualification and validation8 and Chapter 7 on outsourced actions9 that influence each Annex 11 and Chapter 4, additionally proven in Determine 1. We are going to solely talk about related clauses the place relevant. Nevertheless, there’s a must have extra cross reference to relevant elements of EU / PIC/S GMP within the revision of Annex 11 than exists at the moment. For instance, Part 3 of the New Annex 11 is about pharmaceutical high quality system (PQS) however doesn’t point out Chapter 1.10
Naming conference: Clauses of the present Annex 11 might be A11 X and the brand new model as New X.Y. An analogous strategy is taken with Chapter 4: C4.X and New X.
Laws: black, white or fifty shades of gray?
Laws ought to be easy. The brand new draft has 111 clauses7 and is kind of stringent and detailed attempting to keep away from interpretation by regulated customers, whereas the present model is straightforward and desires interpretation.4 Linked carefully with Annex 11 is the replace to Chapter 4, which has ballooned from 32 to 85 clauses.7 These are paradigm adjustments.
Laws ought to observe this strategy:
- Laws ought to outline What needs to be carried out.
- Every firm interprets and decides How the laws might be applied.
- Regulators examine to evaluate compliance with the laws: How = What?
Nevertheless, with the revision of Annex 11 and Chapter 4 there are numerous clauses that state How to conform reasonably than state What ought to be carried out.
Why are regulators attempting to inform us work?
The revision typically mandates comply; this regulatory overreach is illustrated in New 11.5:
… passwords ought to … include a mix of uppercase, lowercase, numbers and symbols.7
This goes above and past what ought to be in a regulation and raises a query: how is that this supposed to extend safety?
Moreover, password complexity is NOT a finest IT observe. Nationwide Institute of Requirements and Know-how (NIST) Particular Publication (SP) 800-63B (2017)11 and the July 2025 replace12 talk about passwords intimately:
- Appendix A Strengths of Passwords: Reviewing password complexity: analyses of breached password databases reveal that the advantage of such guidelines is much less important than initially thought, and the impacts on usability and memorability are extreme.12
- Part 3.1.1.1:
o Passwords SHALL both be chosen by the subscriber (consumer).
o 1. … passwords which might be used as a single-factor authentication mechanism to be a minimal of 15 characters in size.
A consumer ought to have the ability to choose their very own password.
o 5. … SHALL NOT impose different composition guidelines (e.g., requiring
mixtures of various character sorts) for passwords.
o 6. … SHALL NOT require … to alter passwords periodically.
Nevertheless, verifiers SHALL power a change if there may be proof that the authenticator has been compromised.
o The whole password SHALL be topic to comparability, not substrings or phrases that is perhaps contained therein (towards a block checklist).12
See additionally the article on passwords,13 which discusses the 2017 model of NIST SP800-63B.
Utah Medical vs FDA court docket case
That is an instance of a regulator telling an organization qualify tools and validate software program. In 2005, FDA (Meals and Drug Administration) versus Utah Medical court docket case centred on the corporate’s strategy was totally different to FDA’s required strategy. The choose said: Many roads result in Rome.14 FDA asserted that Utah Medical ought to adjust to the 21 CFR 820 laws in a way passable to the FDA. The choose said that laws are:
- … common and versatile in order to cowl a broad spectrum of merchandise and actions, …
Much like the A11. - … have the advantage of generality and the vice of imprecision.14
Not black or white however fifty shades of gray relying on circumstances of a person firm comparable to measurement and the merchandise being manufactured: strong dosage varieties or superior remedy medicinal merchandise (ATMP). That is danger administration in observe, as one measurement not often suits all. - The choose discovered for Utah Medical as the corporate was answerable for their tools and software program, regardless that they didn’t observe what was mandated by FDA.14 For additional particulars, see the article by Burgess and McDowall.15
A lesson learnt?
Is there a lesson that may be discovered from the replace of Annex 1 (Manufacture of Sterile Medicinal Merchandise)?16 This replace, which got here into impact earlier than Annex 11, additionally ballooned in measurement to 59 pages and comprises an excessive amount of element. Though high quality danger administration (QRM) is included firstly into the scope, it doesn’t present the chance for impartial risk-based implementation, which has led to diverse interpretation of “required” language by international well being authorities and by international producers. In observe, some international regulators usually are not amenable to risk-based approaches (e.g., bracketing).
Annex 11 revision seems to be as whether it is going the identical method. Stakeholder suggestions might be essential right here.
Idea paper on the revision of Annex 11
The idea paper on the revision of Annex 116 contained 33 subjects for consideration when updating the regulation and requested trade suggestions. Desk 1 maps the areas within the idea paper towards the clauses in New Annex 11.
Two key omissions are:
- Digitalization and technical controls over handbook processes. Given the transfer to digital transformation below Pharma 4.0,17 it is a missed alternative. This omission is compounded by 21 references to hybrid methods throughout the 2 updates.
- Cloud service suppliers (CSP) usually are not given the visibility they deserve, in distinction with the FDA steering on digital methods, digital information and digital signatures in scientific investigations: questions and solutions,18 EMA guideline on computerised methods and digital information in scientific trials19 and OECD GLP (good laboratory observe) 17 complement 1 on GLP & Cloud Computing.20
Desk 1: Comparability of 2022 Idea Paper with New Annex 11 2025
|
Clause |
Topic |
Annex 11 Clause(s)/Part(s) |
|
1 |
Incorporation of the EMA Annex 11 Q&A |
N/A |
|
2 |
Information in movement / information at relaxation a |
(10.1, 10.2, 10.3) 16 and 17 |
|
Configuration hardening / technical controls |
Not mentioned |
|
|
3 |
Digitalization |
Not mentioned |
|
4 |
No improve in danger when changing a system |
2.8 |
|
5 |
Reference to ICH Q9(R1) |
4.2 |
|
6 |
Cloud service suppliers |
Not mentioned |
|
7 |
Service supplier agreements |
7.5 |
|
Documentation obtainable for inspections |
7.4 and 7.5 |
|
|
8 |
Industrial off the shelf (COTS) software program |
6.1 (partially) |
|
9 |
Defining qualification and validation |
Glossary (poor) |
|
10 |
Give attention to testing GMP performance |
9.6 |
|
11 |
Person necessities and traceability |
6.1, 6.4, 6.5, 9.5 |
|
12 |
Agile software program improvement |
Not mentioned (6.1?) |
|
13 |
Definition of important methods and important informationb |
Not mentioned |
|
14 |
Defend GMP methods, networks and infrastructure and information |
15.1–15.20 and 16.1–16.5 |
|
15 |
Testing information / archived information restores |
16.6, 17.4 and 14.2 |
|
16 |
Backup expectations |
16.1– 16.5 |
|
17 |
Digital copies of knowledge |
12.9 |
|
18–24 |
Audit trails and alarms |
12.1, 12.2,12.3, 12.7, 12.8 and eight |
|
25 |
Configuration assessment |
14.1 and (6.6 partially) |
|
26 |
System and information confidentiality, integrity and availability |
15.1 |
|
27 |
Extra safety controls |
15.1–15.20 |
|
28–30 |
Person authentication and entry privileges |
11.2, 11.3, 11.10 |
|
31 |
Validated archive course ofc |
17.1–17.5 |
|
32 |
AI and ML(machine studying)d |
Not mentioned |
|
33 |
Take into account enter from FDA draft pc software program assurance (CSA) Steerage |
Not mentioned |
a For disposal of information see chapter 4.79
b Integrated in chapter 4.13
c Additionally thought of in chapter 4.76
d See Annex 22 (out of scope of this text)
Gone, however not forgotten?
In New Annex 11, present key necessities from the present model have been omitted:
- A11 Precept: … IT infrastructure ought to be certified.
That is an abysmal omission as questions have been raised already: as this has been omitted from the draft, given the prescriptive nature of the doc, I don’t must qualify my infrastructure anymore? That is equal to constructing a home with out foundations; see our feedback below part 2. - A11 3.4 Audit info of suppliers
Availability of provider high quality system and audit info ought to be made obtainable to inspectors on request is now not in New Annex 11. - A11 4.3 Stock and system description
The stock of methods and system description for important methods are omitted – why? These are important omissions, as you want a listing of methods for asset administration in addition to regulatory compliance. After a system danger evaluation, ALL computerized methods might be listed in an up-to-date stock in order that objects which might be non-GMP tools, devices or methods in a GMP-area are correctly recognized and labelled Not for GMP use. - A11 4.7 Automated check instruments
… Automated testing instruments and check environments ought to have documented assessments for his or her adequacy. Given the rise of validation administration instruments since 2011, this paragraph ought to have been expanded and enhanced, not dropped. It additionally gives justification for not validating automated check instruments, as some suppliers state these have to be validated, however documenting them for adequacy.
o There’s a point out of utilizing a instrument for traceability in New 6.5 however it’s inadequate given the present availability of lifecycle instruments.
o Nevertheless, out of nowhere in New 4.23 and 4.25 there are mentions of computerized validation scripts,7 these usually are not equal to the usage of automated check instruments.
o There have to be a piece discussing regulatory necessities about instruments supporting GxP/GMP actions comparable to automated testing instruments and validation administration methods nevertheless it have to be in Annex 11 not in Chapter 4.
- A11 4.8 Information migration
If information are transferred to a different information format or system, validation ought to embrace checks that information usually are not altered in worth and/or which means throughout this migration course of.4
New 10.3 has a clause on information migration, however doesn’t cowl the alteration in worth and/or which means. - A11 8 Printouts
These are now not required throughout an inspection, the emphasis is on e-records. A change for the great. - A11 13 Incident administration
IT incidents and issues are key inputs for periodic critiques. Once more, one other important omission from the revision. - A11 15 Batch launch
Launch of batches by a certified individual (QP) utilizing an digital signature is omitted however that is lined in Annex 16.21 In New Annex 11, it’s changed by a requirement to permit a QP entry to audit path entries. Good luck with that! A QP ought to at all times have entry to all information and knowledge related to the batch, the place vital they carry out a assessment with the assistance of skilled(s). - Glossary
Course of and system house owners are deleted: this avoids allocation of tasks. Moreover, IT incidents and issues usually are not outlined nor are IT adjustments. Glossaries are mentioned later on this article.
Regulatory plagiarism?
It’s déjà vu once more with a few of the clauses in each Annex 11 and Chapter 4. The place have they arrive from? See Determine 2.
- An info safety administration system (ISMS) below ISO 27001 turns up once more in New 15.1. An upper-case ISMS was proposed within the 2008 replace of Annex 113 and rejected by the trade however makes an unwelcome lower-case return.7 Second time fortunate? Mentioning this means that an ISO 27001 ISMS22 ought to be applied and licensed, which can be not possible for small regulated customers.
- PIC/S PI-04123 gives enter to many sections to Chapter 4. This isn’t stunning as part 3.7 of PIC/S PI-041 states This information isn’t necessary or enforceable below regulation.23 What you see is a distillation of key elements of PIC/S PI-041 steering into regulation, e.g., management of clean varieties is in steering paperwork and is included in Chapter 4 to grow to be a regulation.7 Ideally, it is advisable to digitalize processes to eliminate paper.
- WHO’s TRS 1033 Annex 4 DI steering from 202124 gives some enter to New Chapter 4.
- The most important copy and paste supply is from a GLP doc: OECD GLP 25 on IT safety,25 revealed in late 2024, which will be mapped fully or partly to 10 clauses in Annex 11 Part 15 and 5 clauses of Part 16, that are copied both verbatim or with minor modifications (see Desk 2 for the instance for anti-virus software program). Contemplating the totally different context inside GxP, the query is whether or not it’s useful to push and combine GLP necessities and definitions into GMP? The reply is exterior of our dialogue on this article.
- OECD GLP 17 on Computerised Techniques is the verbatim supply for 9 of the entries within the New Annex 11 glossary.26 A word to the authors, copy and paste is nice however the definition for COTS software program nonetheless comprises the GLP time period check facility administration (TFM). Doh!
The issue with COTS is that the time period isn’t per GAMP 5 SE.27 We are going to talk about COTS later on this article.
Determine 2: Enter sources to the proposed revision of Annex 11 and Chapter 4. Credit score: Bob McDowall.
Desk 2: Sections on anti-virus software program from OECD GLP No 25 and New Annex 11
|
OECD GLP 25 |
New Annex 11 |
|
9 Anti-virus software program ought to be put in and activated on methods utilized in GLP, as acceptable. The anti-virus software program ought to be repeatedly up to date with the latest virus definitions so as to establish, quarantine, and take away recognized pc viruses. This course of ought to be monitored. |
15.18 Anti-virus software program ought to be put in and activated on methods utilized in GMP actions, particularly these interfacing the web. The anti-virus software program ought to be repeatedly up to date with the latest virus definitions to establish, quarantine, and take away recognized pc viruses. The effectiveness of the course of ought to be monitored |
Observe: phrases not matching are in italic font and underlined.
Part 2 precept
As talked about earlier, the next ten phrases have to be retained on this part:
The applying ought to be validated; IT infrastructure ought to be certified.
This clearly encapsulates the essence and one of many key necessities of Annex 11. Omission within the launched model can be an abrogation of obligation by EMA and PIC/S.
- 2.2 High quality danger administration
Part 4 is on QRM and refers to ICH Q9 (R1).28 QRM is repeated in New 4.3, 6.2, 7.2 and 9.2, which agree that the extent of efforts for system validation ought to be outlined based mostly on the system danger evaluation and the supposed use. QRM is parroted all through each revisions when all that’s required is a single point out. - 2.4. Information integrity
DI is mentioned in sections 10, 11, 12 and 13. The enlargement is to Safety and information retention.
New 2.4 mentions ALCOA+, whereas the proposed revision of Annex 11 lacks the dialogue of this precept. There may be additionally a foul day on the coordination workplace as Annex 11 mentions ALCOA+ and New 4.63 ALCOA++ with the emphasis on traceability, just like the replace to USP <1029> on Good Documentation Tips and Information Integrity.29 This disconnect is carried by way of to the Glossaries in each paperwork, as we critically assessment later. - 2.6. Outsourced actions
Part 7 explains the qualification necessities for vendor, service supplier and inner IT, nonetheless it lacks any clause on cloud providers. Use of cloud was a motive for the Annex 11 replace, as said within the idea paper and the New Annex 11.6,7 - 2.8. No danger improve
The assertion within the New Annex 11, The place a computerised system replaces a handbook operation, … There ought to be no improve within the total danger of the method, is prolonged to incorporate alternative of 1 system by one other. That is the second key requirement of Annex 11 since 1992. Why has it been relegated to the again part of the precept, as a substitute of preserving it as a foremost precept?
The precept will be condensed drastically as a result of a lot of the content material is repeated later within the doc.
Part 3 pharmaceutical high quality system (PQS)
Why repeat what’s already in Chapter 1 PQS10 and Chapter 9 self-inspections30 in clauses 3.1 i and three.1 iii? Why create repetition and redundancy? Cross reference can be higher.
Clause 3.1 ii covers adjustments to a computerized system, nonetheless A11 10 has a separate part and heading which highlights the significance of Change and Configuration Administration which is hidden within the replace.
A very good level is the reinforcement of the function of administration within the implementation and operation of computerized methods in New 3.1 iv and v.
BUT, there isn’t any level having administration in control of computerized methods in the event that they don’t perceive them. Tender Company and Stason Pharma have been cited in FDA warning letters31,32 for lack of senior administration and high quality assurance oversight of computerized methods, for additional studying see the evaluation of those two warning letters.33
We recommend incorporating these in part 5 personnel and coaching however reverse the order of those two sections to reinforce the tasks of administration and their coaching.
Part 4 danger administration
A11 1 on danger administration may be very easy: apply danger administration all through the lifecycle.4 This may be interpreted as vital relying on the chance posed by the system and the required controls to make sure the integrity of the info.
In part 4, we’ve got 5 clauses going into element on lifecycle identification and evaluation, acceptable validation, mitigation and DI. As well as, 4.5 of New Annex 11 mentioned information vulnerability below DI. Our suggestion is to rephrase the subtitle into Information danger to remain per New Chapter 4 (4.13) and PIC/S PI-041 part 5.5.23 Alternatively, file danger is a greater phrase. As mentioned earlier, QRM or danger administration is referred to 10 occasions in New Annex 11, which is an excessive amount of and too detailed.
Take A11 clause 14 and add enterprise functionality or course of understanding because the fourth danger criterion and reference to ICH Q9(R1)28 is all that’s required.
Part 5 personnel and coaching
New 5.2 talked about … ample system particular coaching … Together with the GMP consciousness, coaching on information safety rules is a requirement as said in New 15.3 and is per part 5.3.2 of OECD GLP 17 complement 1.20
Nevertheless, there isn’t any point out of any evaluation of competence or understanding. Competence must be ensured by totally different selections, e.g., utilizing a questionnaire34 or simulated checks for cybersecurity (New 15.3). Once more, the latter choice in New 15.3 centred on How to do, not What to be achieved.
To seek out any coaching evaluation, it is advisable to go to New 4.45 (vi) … verification of the effectiveness of coaching. Nevertheless, EU / PIC/S GMP Chapter 2 clause 2.11 on coaching states that its sensible effectiveness ought to be periodically assessed.35
The 2 clauses on the function of administration ought to be positioned right here, as talked about in Part 3 of this text. Don’t neglect that administration want coaching too, particularly on the implementation and validation of computerized methods!
Part 6 system necessities
A11 4.4 merely states:
Person Necessities Specs (URS) ought to describe the required features of the computerised system and be based mostly on documented danger evaluation and GMP influence. Person necessities ought to be traceable all through the life-cycle.4
A easy and straightforward to grasp clause: you want a URS and any additional specification paperwork to adequately outline the supposed use of the system. A URS is crucial doc in any validation undertaking because it defines the supposed use; no URS, no validation. Necessities ought to be traceable – the technique of how to do that is left open.
Nevertheless, each the prevailing and New part 6 don’t state supposed use coupled with underlying course of understanding and, the place acceptable, enchancment.
New Annex 11 has 6 clauses to explain doc consumer necessities, utility configuration and mandates a traceability matrix. All you want is a approach to show traceability – how that is achieved is as much as a regulated consumer. Traceability from the URS must also embrace SOPs, provider evaluation, set up and commissioning by a provider in addition to a validation abstract report (VSR).
A very good VSR is important as it’s the place to begin for regulatory inspections as per clause 23.10 of PIC/S PI-011.36 BUT a VSR isn’t outlined in GMP laws. Part 9 of New Annex 11 is poor as there isn’t any formal mechanism for system launch discussing the general validation – warts and all – by not concealing any issues encountered in a undertaking. Due to this fact, a VSR ought to be outlined in Annex 11.
A very good level that has been added is that consumer necessities reside paperwork and have to be up to date however model managed (New 6.4). As data of the system develops or extra features are used, the URS and different specs have to be up to date, with consequential knock on to different validation paperwork as vital, e.g., check scripts.
Configuration of an utility is essential and have to be prospectively specified and documented in a separate configuration specification (New 6.6), relying on system complexity. Nevertheless, the issue with New Annex 11 is that configuration can refer to 2 areas:
- Software configuration: the technical management settings to make sure DI and information safety, e.g., definition of consumer roles with entry privileges.
- System configuration: an inventory of all elements of a system, comparable to digital or bodily pc system, information storage, peripherals, working system, utilities, middleware, database and utility.
Each are important for change management. Nevertheless, the Glossary of the New Annex 11 solely refers to system configuration below the time period configuration.7
Part 7 provider and repair administration
That is expanded from A11 3 and now consists of 5 clauses. How a lot overlap is there with Chapter 7 on outsourcing? 9 A very good level in New 7.1 is that the duty and accountability stays with the regulated consumer of service suppliers, together with the corporate’s IT Division. New 9.9 reiterates this level below Authorisation.
Provider evaluation is now an audit or a radical evaluation in New 7.2, reasonably than a necessity for an audit ought to be based mostly on a danger evaluation. It seems that evaluation questionnaires are consigned to the spherical submitting cupboard of historical past as ineffective? See the article on number of a CSP37 to separate clouds from clods.
Necessities for contracts are very detailed in New 7.5 and seems as a “how-to write a contract” instruction. There may be not a particular or detailed part on cloud computing which is barely talked about in passing.
There’s a disconnect in New part 7 relating to the requirement on service degree agreements (SLA):
· New 7.3 describes the extent of oversight by way of defining SLA and key efficiency indicators (KPI) for service supplier or an inner IT division,
· New 7.5 mentions … the regulated consumer ought to have a contract with a service supplier or have accredited procedures with an inner IT division.
New 7.5 (Contracts) – ought to this degree of element be in a regulation? If it stays, the clause ought to be re-ordered for higher understanding: duty, audit, contracts, documentation availability and oversight. For extra element on the scope, together with use of subcontractors by a provider and content material of SLA, see our full article.38
There may be nothing intimately on the cloud and the function of a SaaS (Software program as a Service) provider in aiding inspections. Higher sources of regulatory steering are:
- Part C on info know-how service suppliers and providers sections of the FDA steering for digital methods, digital information, and digital signatures in scientific investigations. 18 This steering additionally gives easier and simpler to learn sections on each utility and IT providers compliance.
- Part 6.7 and Annex 1 on cloud options and agreements within the EMA guideline on computerised methods and digital information in scientific trials. 19
- OECD GLP 17 Complement 1 on GLP and cloud.20
Maybe again to the longer term is named for by retaining IT-departments ought to be thought of analogous within the present 3.1 and equating it to an exterior service supplier?
Part 9 qualification and validation
How about this for one more unhealthy day on the workplace? Annex 11 says observe Annex 15 for qualification and validation and Annex 158 says observe Annex 11 for validation of computerized methods, as proven in Desk 3. A regulatory Groundhog Day? Who has not learn Annex 15?
Desk 3: Cross-reference confusion between Annexes 11 and 15
|
New Annex 11 |
Present Annex 15 |
|
9.1 Ideas. Qualification and validation actions for computerised methods ought to observe the overall rules outlined in GMP Annex 15. |
Precept: Computerised methods used for the manufacture of medicinal merchandise must also be validated in accordance to the necessities of Annex 11. |
Even in case you observe the qualification and validation part in Annex 15, we meet the 4Qs mannequin which ought to have been pensioned off way back. FDA discontinued the use in 2002 with the overall rules of software program validation39 and GAMP 5 stopped utilizing 4Qs in 2008.40 Additionally Annex 15 mentions manufacturing unit acceptance checks (FAT) and website acceptance checks (SAT) that are usually relevant to manufacturing methods.8
The separation of specs in New part 6 from the rest of the lifecycle is unusual: sections 6 and 9 ought to be built-in collectively, as it’s within the present Annex 11, and are available after part 7, e.g., assess provider(s), specify and validate the system.
New 9.4 check proof ought to observe GAMP 5 SE, stating major check proof and secondary check proof: use the audit path to doc testing and solely use display dumps when vital, e.g., to hint the actions again to the necessities.27
This part solely considers the execution of check scripts. Nevertheless, important necessities are additionally verified by way of writing SOPs for system use, execution of provider set up and commissioning (set up qualification (IQ)/ operational qualification (OQ)) protocols, which aren’t lined. Written directions (e.g., SOP) are additionally required to carry out the validation. Extra particulars on Procedures will be present in New 4.27.
New 9.7 Validation plan ought to be established after system choice and the influence/danger evaluation. It’s formal proof of validation that’s within the scope of audit and inspection. Our suggestion is to debate it below 9.4.
CSA is lifeless as a dodo!
FDA draft CSA steering41 allowed the usage of unscripted testing for medical machine high quality system and manufacturing software program. Because of this, many have jumped on the bandwagon of unscripted testing to be utilized to Pharma.
New 9.4 and 9.7 state:
System qualification and validation ought to present proof within the type of executed check scripts …
… Take a look at scripts ought to be described in adequate element to make sure an accurate and repeatable conduct of check steps and stipulations.
The requirement for check scripts to be described in adequate element for … repeatable conduct is an entire rejection of CSA unscripted testing.
Nevertheless, there ought to be emphasis on utilizing skilled customers to put in writing and execute check scripts.
How a lot element is ample? That is within the eye of the inspector however commensurate with the system danger.
A check script should include adequate element to execute directions together with acceptance standards which might be clear and concise. In a laboratory, a skilled analyst is aware of what to do when given an instruction in an analytical process: put together 1L 0.1M sodium acetate answer. This entails:
- Calculating the burden of sodium acetate vital
- Checking the analytical stability is certified and works inside limits
- Weighing the fabric
- Transferring it to a volumetric flask
- Dissolving the strong and making as much as quantity
- Labeling the storage vessel with contents, identify of analyst and dates or preparation and expiry
- All work being recorded as vital with materials identify, expiry date and batch quantity, stability printout, and many others.
Equally, a check script mustn’t require each mouse click on or menu choice to be specified and examined. Skilled customers usually are not idiots; laws mustn’t deal with them as such.
Part 10 information dealing with
A number of the new necessities are just like present A11 clauses.
- New 10.1 verification of handbook information entry equates to A11 6 (Accuracy Checks)
- New 10.2 (Information switch) equates to A11 5 (Information).
Information migration is mentioned twice in New Annex 11:
- New 10.3 signifies … The place an advert hoc course of requires that important information or an entire database be migrated from one system to a different …
- New 15.11 states Validation of purposes on up to date working methods and platforms and migration of knowledge ought to be deliberate and accomplished in due time previous to the expiry of the seller’s help.
From expertise, information migration isn’t an advert hoc course of. NEVER undertake an advert hoc migration; information migration must be rigorously deliberate. The supply and vacation spot methods and the way in which each shops information have to be totally understood. A case research of knowledge migration will be discovered within the guide on validation of chromatography information methods (CDS). 42 C4.10 and New 4.76 each point out controls are required to make sure the integrity of the file all through the lifecycle.5,7,43
Part 11 id and entry administration
From 10 phrases in A11 12.3, we now have one other instance of regulatory bloat to 11 clauses and prescription of what to do. Aside from the detailed requirement in New 11.5 (safe passwords) that was mentioned earlier on this article, we’ve got the next:
- New 11.1 states … Using shared accounts apart from these restricted to read-only entry (no information or settings will be modified), represent a violation of knowledge integrity.
Using shared read-only accounts is a poor IT observe. If present in an audit, there may be at all times a query: are there extra severe examples of account sharing? - New 11.6 gives one other instance of claiming do as a substitute of what to be carried out. Multifactor authentication (MFA) is without doubt one of the doable technical choices (immediately) however will not be helpful sooner or later.
- New 11.8 on inactivity logout is in keeping with PIC/S PI-041 9.5. no 123 Techniques ought to embrace an computerized inactivity logout, which logs out a consumer after an outlined interval of inactivity …
An automatic inactivity display lock can current a significant downside in an analytical laboratory. Many laboratory methods are standalone and can run unattended in a single day. If an inactivity logout is used, methods might cease working until there’s a performance to allow a consumer to sign off and this permits the sequence to nonetheless function.
On this part, there are additionally good necessities for:
- Distinctive accounts
- Confidential passwords
- Segregation of duties
- Least privilege precept
- Checks to make sure that consumer accounts are nonetheless required
Nevertheless, you need to have been doing this already!
One level that’s missed on this part is in regards to the system’s means to generate an inventory of customers and their consumer roles / entry privileges as mentioned in PIC/S PI-041 9.5. 23 This can be a useful requirement that ought to normally be addressed in a URS.
Part 12 audit path (AT)
There are two updates, that are constructive:
- Opinions. It’s important to have an SOP for every system AT assessment. That is per our latest AT article44 however is opposite to PIC/S PI-041, part 9.8 no 123 that solely requires a single AT assessment SOP.
- Digital copy. Digital AT entries at the moment are required as a substitute of a printout.
Nevertheless, there are poorly written clauses:
- The primary clause on consumer interactions is insufficient, as an AT should seize system actions as nicely. Think about you probably have an in a single day run in a laboratory: You begin the sequence, which is logged in an AT. You go residence and the remainder of the work is captured routinely; no handbook involvement, so no AT entries?
Each operator interplay and system entries are producing GMP information and because the glossary states, AT permits reconstruction of the occasions so we must always have the same AT performance for system and consumer interactions.44 - What about computerized information switch? There ought to be an entry within the supply system AT (e.g., CDS) and a corresponding one within the vacation spot system AT (e.g., laboratory info administration system).
- New 12.3 No edit or deactivation: AT have to be configured and activated at set up and can’t be turned off (good). Idea paper merchandise 18 states AT performance is necessary and any grace interval has lengthy expired,6 however New part 12 fails to say it explicitly.
o The flexibility to deactivate an AT by a system administrator is completely unacceptable and have to be deleted from this regulation.
- New 12.4 lacks dialogue of assessment by exception talked about in part 9.6 no 1 of PIC/S-041.23
- An choice to Export of the info to a instrument is sluggish and if the export is textual content, it will increase the chance of DI violation.
- The clause on Unbiased assessment wants clarification that the originating division should carry out AT assessment as said by PIC/S PI-041 part 9.6 no 123 and the FDA DI information, Q7.45 As well as, Q16 asks Ought to personnel be skilled in stopping and detecting information integrity points as a part of a routine CGMP coaching program? Sure.45
- New 12.7 omits the criticality of knowledge and metadata evaluation that should be reviewed.
- Availability to QP. Good luck with this one! Most likely, a QP will need assistance from a topic skilled (SME) as more than likely they aren’t a consumer of the system. What in regards to the different e-records on a system, are they off limits?
Part 13 Digital signatures
An earlier article by certainly one of us in contrast the e-signature necessities for A11 14 with these in 21 CFR 11 and questioned why the FDA required 640 phrases (not together with definitions) to realize what Annex 11 achieved in 40 phrases?46 This was the facility of decoding the phrase digital signatures … have the identical influence as hand-written signatures throughout the boundaries of the corporate.4
The proposed model now has 9 clauses and 357 phrases. Solely one other 283 phrases to meet up with the FDA!
This part have to be learn along side clauses New 4.64–4.75 on signatures in GMP related paperwork.
New 13.6 Manifestation: there are variations between New Annex 11, 21 CFR 11 and even New Chapter 4 (see Desk 4).
Desk 4: Comparability of digital signature necessities of 21 CFR 11, New Annex 11 and New Chapter 4
|
New Annex 11 (13.6) |
21 CFR 11.50(a) |
New Chapter 4 |
|
|
|
The additions of consumer id and the function of the signer add zero worth to an digital signature. New 4.73 agrees that the which means of an e-signature implies the function of the signer.
Additional comparability is about New 13.4 and New 4.65, indicating the usage of date and time. The query is that if date and time is necessary with a signature on a paper GMP file? For an digital signature, sure, it’s, as a result of it routinely timed and dated, nonetheless for handwritten signature it might be date solely. Why signal a file with time on paper? That is an over burdensome and pointless requirement.
New 13.8 Unbreakable hyperlink Digital signatures ought to be completely linked to their respective information. Controls ought to be in place to make sure that a signed file can’t be modified or alternatively, that if a later change is made to a signed file, it’ll clearly seem as unsigned.
- The primary sentence is okay, however the underlying e-record set have to be locked and can’t be modified.
- What 21 CFR 1147 or New 13.8 each omit is the problem of revoking an e-signature. Within the cellulose world, if an accredited report is discovered to have a mistake, the report is recalled, the error corrected, accredited and reissued. The doc historical past will file the replace and the unique report will nonetheless be obtainable as an archived model.
- If a signature has been eliminated and the file is now unsigned might this be used as a method for falsifying information?
- Eradicating an e-signature from an digital file compromises ALCOA++ standards. The e-signature removing signifies that a file isn’t full, not correct and work is actually not traceable.
- In an digital world, you want a perform to revoke digital signature(s); the signatures nonetheless be seen on the report, the rationale for revoking have to be documented on the file and within the AT, e.g., a mistake ought to be defined, comparable to a miscalculation. When corrected, the report is re-signed electronically and reissued. The entire signing sequence: unique e-signatures, revoking e-signatures and e-resigning should all be obtainable to make sure traceability (the tenth ALCOA++ criterion – however clearly solely in New Chapter 4).
New 13.9 might be mentioned later below hybrid methods.
Signatures in GMP documentation: attribution versus signing
Almost 30 years in the past, following the publication of 21 CFR 11 for digital information and digital signatures, 47 we had a debate and resolved the distinction between attribution of motion versus signing of a file in a computerized system. There are only a few necessities for signing information: reporting ultimate check outcomes, certificates of study, qualification and validation paperwork, technique validation protocols and experiences, and many others. Nevertheless, sections on signatures in New 4.64–4.75 and Part 13 in New Annex 11 reopen that debate, as there isn’t any said separation between attribution and signing, solely the requirement for signatures.
It seems at first sight all the pieces have to be signed, however lastly in New 4.68 the regulated consumer ought to have recognized these information which require a legally binding signature. This ought to be the primary requirement on this part.
New 4.64 Signatures are important for guaranteeing accountability …
Does each motion on paper or utilizing a computerized system now require a signature? No, identification of a person performing an motion is the important thing goal.
There are numerous superfluous necessities in clauses 64–65 of New Chapter 4.
Use of initials, widespread in some international locations, requires a process to allow this. In EU GMP Chapter 6 (High quality Management) there are solely necessities for initials for testers and reviewers in clause 6.17 (f) and (g) and the one signature requirement is for the batch launch in clause (h).48
Part 14 periodic assessment (PR)
The replace is extra detailed which is sweet; overlaying not simply guaranteeing the validation standing of the system but in addition the usage of it, e.g., verify AT assessment effectiveness. New 14.1 requires a report containing findings and corrective actions, which might be checked within the subsequent PR of the system as said in New 14.2 iv.
New 14.3 introduces risk-based frequency of PR contemplating system criticality and information danger and vulnerability so the frequency of PR will fluctuate between methods. That is essential as many organizations push a PR for all methods to a few years – that is lazy and mistaken, as some methods are larger danger than others. We recommend that for important methods, a PR six months after implementation ought to be good observe to make sure all is working appropriately, as ready two to a few years can be too late to right any issues. The problem of frequent updates below SaaS settlement isn’t addressed on this part. For sensible info and recommendation on PR, see.42
Part 15 safety
Oh pricey, it is a procuring checklist of 20 clauses that’s brutally prescriptive and shouldn’t have any place in a regulation. This raises the query of verification by inspectors, who usually haven’t any experience on this space. As famous in Determine 2, ten sections have been lovingly ripped off from OECD GLP 25.25 These 20 duties are what any competent IT division or service supplier ought to be enterprise already.
New 15.2 Steady ought to be continuous to be per GMP Chapter 1.10 Continuous is a discontinuous course of that permits for change management, which is important for guaranteeing compliance. Chances are you’ll be assessing cybersecurity threats repeatedly however updating methods frequently.
Catastrophe restoration (DR) plan is described in New 15.7; nonetheless, it’s inadequate in addressing DR parts:
- Each restoration level goal (RPO) (how a lot information are you able to afford to lose) and restoration time goal (RTO) (the time it takes you to recuperate the info or system) have to be outlined in a DR plan.
Why does New 15.7 solely contemplate RTO? - Enterprise continuity (BC) is centred on various working preparations. 27 Nevertheless, with growing digitalization of Pharma, various working practices based mostly on paper are inadequate, due to this fact sustaining computerized system operations is important and requires failover in separate areas (see New 15.6 Replication).
- DR and again up (part 16) are interconnected: with out again up there isn’t any DR.
- New 15.15–15.17 permit the usage of USB (common serial bus) units. These have main safety and information vulnerability points and ought to be averted completely.49
Part 16 backup
Backup is expanded from one to 6 clauses within the new model based mostly on OECD GLP 2525: a lot of that is widespread sense and begs the query, why ought to it’s in a regulation? If this degree of element is required, then why not add a clause requiring a backup of knowledge, utility and system earlier than making a change? In case of issues, you already know it is sensible.
Replication can also be mentioned in New 15.6 however that is a part of an total backup technique, given the dependency of Pharma on computerized methods.
A11 7.2 states … Integrity and accuracy of back-up information and the power to revive the info ought to be checked throughout validation and monitored periodically. The readability and ease of this sentence is misplaced in New 16.6 because it implicitly mentions … examined and documented based mostly on danger throughout system validation …
The content material in New 16.1–16.4 can also be repeated in New 4.76.
Part 17 archiving
The next combines our feedback on part 17 and New 4.76–4.79 for digital information.
Somewhat than archive laboratory information off-line, it could be higher to archive within the system that created the information. This has the benefit that each archived and dwell information will be up to date if vital and avoids the necessity to verify the readability of archived information. Nevertheless, this can rely upon the storage obtainable, ample again up with fail-over and if features in an utility allow archived information to be locked and saved safe. See OECD GLP 15 part 8 for a greater dialogue of an digital archive.50
An alternate viewpoint is NIST SP800-209 for Safety Issues for Storage Infrastructure, which suggests:
IS-SS-R1 (b) Lengthy-term archive and backup methods ought to be separated from manufacturing information storage methods.51
There could also be archive on the manufacturing system however it’s crucial that there are restoration copies on separate off-line and off-site storage. No matter the place information are saved, a regulated consumer should guarantee long-term availability of knowledge.
One challenge that isn’t talked about in New 4.79 on keep away from destroying the mistaken information on the finish of the info lifecycle, is that if there may be any authorized motion pending? While not a regulatory challenge, it’s a authorized one and a few information might require a authorized maintain to forestall destruction even when the retention interval has expired.
New 4.26 reiterates that dynamic information shouldn’t be transformed to static information; it is a good level and is per present steering paperwork.23,45,52
Retention of paperwork: definition of location
The potential downside with each New Annex 11 (16.4) and Chapter 4 (4.76) is that there isn’t any definition of location, which is analogous in GLP laws.49,53,54 Location of paper information is easy both in a constructing on website or in a doc repository: location is the road handle. Nevertheless, what’s the state of affairs if utilizing a CSP? Will a uniform useful resource locator (URL) be ample or do you want the handle of the info centre? This was the query posed in our latest article on SLA38 and in addition in part 6.3 of OECD GLP 17 complement 1.20
Many web service suppliers (ISPs) is not going to present the handle of their information centre as it is a safety breach. Regulators should convey their laws into the true world and resolve if a URL is an ample location for entry to a cloud repository.
Chapter 4 documentation and its influence on Annex 11
The present Chapter 4 is comparatively concise with a precept and 32 clauses. It has expanded drastically within the revision to a precept and 85 clauses. This chapter is dedicated to documentation, nonetheless if the GMP documentation is saved as a set of digital information, the necessities of Annex 11 have to be utilized and there’s no want to debate digital matter once more in Chapter 4. Why repeat Annex 11 necessities in Chapter 4? This causes confusion and makes efficient interpretation (the place allowed) way more tough.
Precept: a important lacking phrase
The Precept of the present Chapter 4 begins with Good documentation, to set expectations for the reader. 5 New Chapter 4 goes downhill instantly by simply stating Documentation. Readers should wait till New 4.56 for a point out of Good documentation.7 Why delete a important phrase on the very begin of Chapter 4 and set a low expectation in contrast with the present model?
New 4.8 makes clear that every one documentation, no matter creation in-house or outsourced, should meet GMP necessities for legibility, accuracy, integrity and completeness.
Hybrid methods in Chapter 4?
Hybrid methods weren’t mentioned within the idea paper for Annex 11.
6
The 2016 WHO TRS 996 Annex 5 comprises the phrases
55
:
- Using hybrid methods is discouraged
- Substitute of hybrid methods ought to be a precedence
The identical strategy is taken by PIC/S PI-041.23 To cite that eminent GxP skilled, William Shakespeare: To hybrid or to not hybrid, that’s the query!
But after we come to New Annex 11 and Chapter 4, hybrid methods are talked about 19 occasions in New Chapter 4 and twice in New Annex 11. The closest we come to digitalization within the two updates is New 4.70:
If information exist electronically such information ought to be signed electronically. Using a hybrid system ought to be averted.
If signatures exist parallel in paper and electronically (e.g., in hybrid methods), the regulatory related signature ought to be outlined by the regulated consumer.7
The primary sentence is nice, supplied that the system has digital signature functionality and, extra importantly, a regulated consumer implements, validates and makes use of it. The latter is usually the rate-limiting step; paper trumps e-records in lots of organizations. The issue is that there’s extra info in an digital file than will be discovered with a printout. This autumn of the information and experiences degree 2 steering on the FDA web site,56 Q10 within the FDA DI steering45 and OECD GLP 17 clause 10726 all point out digital information is the true GMP file as they include extra info than paper printouts.
The 4 clauses for hybrid methods in New 4.82–4.85 require:
- Threat-based validation and management however hybrid methods current excessive DI danger anyway.
- An in depth description of the whole system. Why do hybrid methods solely require a system description, particularly when this has been dropped from Annex 11?
- The interface between handbook and the computerized system have to be managed and managed with but extra QRM. That is merely restating the clause on accuracy checks in A11 6 utilizing extra opaque and wordy language.
- Analysis, approval and archiving of knowledge of each e-records and paper require procedures.
What’s the level of those clauses after they merely restate what’s already in Annex 11?
The signature requirement in New 13.9 for hybrid methods presents a technical problem for suppliers and controlled customers alike:
13.9. Hybrid answer. If a wet-ink signature … is used to signal digital information held in a computerised system (a hybrid answer), measures ought to be applied to offer a excessive diploma of certainty that any change to the digital file will invalidate the signature. This can be applied by calculating a hash code (verify sum) of the digital file and printing that on the signature web page.7
- What is that this doing in a piece on digital signatures when it ought to be in Chapter 4 or a dustbin? It might be a lot better if the laws persuaded regulated customers to keep away from utilizing hybrid methods as advocated by WHO and PIC/S steering paperwork.23,55
- It’s inappropriate and a prescriptive regulatory overreach.
- Presently, it’s unlikely that any hybrid system might meet this requirement.
- It’s extremely unlikely that suppliers would develop such a performance.
- If applied within the ultimate launch of Annex 11, all hybrid methods are out of compliance instantly.
Glossary issues, omissions and inconsistencies
There are issues with glossaries, not simply with the New Annex 11 and Chapter 4,7 but in addition with A114 glossary and EU GMP glossary.57 Comparability of chosen entries throughout all 4 glossaries is proven in Desk 5.
Extra worrying are the omissions. For instance, there isn’t any definition of deviation in any EU GMP glossary.
Desk 5: Annex 11, Chapter 4 and EU GMP Glossary consistencies and inconsistencies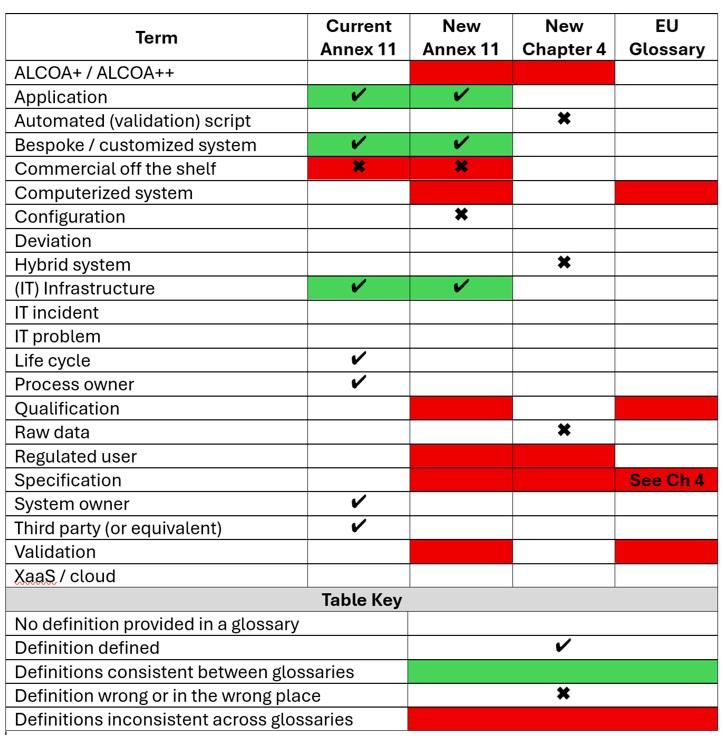
Glossary omissions
The next phrases in A114 have been omitted from the revised model and should be included within the ultimate model of the replace:
- Course of proprietor
- System proprietor
- Third get together
- Lifecycle: At the least this must be outlined on a common degree overlaying computerized methods.
Undefined phrases
An IT incident or downside is normally not a deviation however many QA departments classify them as such. That is regardless of the wording within the present and proposed revision of Annex 11 in PR: incidents, issues and deviations are separate objects.4,7 An IT incident, comparable to a backup failure, normally has no influence on product high quality or affected person security and due to this fact shouldn’t be categorised as a deviation. Nevertheless, if an incident isn’t dealt with in a well timed and ample method, this might result in a deviation.
Any definition of cloud providers has been omitted from the Annex 11 glossary. Given the usage of cloud in Pharma it is a important omission.
Uncooked information definition is mistaken
Uncooked information is a GLP time period that has been obtainable since 1978 in 21 CFR 58.3(ok):
Uncooked information means any … information … or precise copies thereof, which might be the results of unique observations and actions of a nonclinical laboratory research and are vital for the reconstruction and analysis of the report of that research …53
The inclusion of uncooked information into EU GMP started in 2011 with the present model of Chapter 45 however with no definition. The MHRA 2018 GXP DI steering outlined uncooked information as:
Uncooked information is outlined as the unique file (information) which will be described because the first-capture of data …52
The reason, however not the definition, then states Uncooked information should allow full reconstruction of the actions.52 Nevertheless, solely the MHRA definition was copied into PIC/S PI-041 as:
Uncooked information is outlined as the unique file (information) which will be described as the primary seize of data, … 23
Nevertheless, the reconstruction of the report has been misplaced. This error is then transferred to the up to date Chapter 4 definition of uncooked information.7
A greater strategy can be to make use of the phrase full information from 21 CFR 211.194(a).58 Additional studying on this topic will be discovered right here.59,60
Glossary inconsistencies
Desk 6: Glossary inconsistencies
|
Time period |
Definition 1 |
Definition 2 |
|
ALCOA |
ALCOA+ |
ALCOA++ |
|
Computerised System |
A computerised system is a perform (course of or operation) built-in with a pc system and carried out by skilled personnel. The perform is managed by the pc system. The controlling pc system is comprised of {hardware} and software program. The managed perform is comprised of apparatus to be managed and working procedures carried out by personnel. |
A system together with the enter of information, digital processing and the output of data for use both for reporting or computerized management. EU Glossary57 |
|
Qualification |
Motion of verifying that the system (together with {hardware} and software program) is successfully designed, put in, commissioned, and operates appropriately. Consult with Pc System Validation New Annex 117 |
Motion of proving that any tools works appropriately and really results in the anticipated outcomes. The phrase validation is typically widened to include the idea of qualification |
|
Regulated Person |
An organization regulated below GMP |
Advertising Authorisation Holder, Producers, management laboratories, importers, and wholesale distributors (if the wholesale distributor holds a producing license) |
|
Specification |
A doc that specifies, in a full, exact, verifiable method, the necessities, design, behaviour, or different traits of a system or element, and infrequently, the procedures for figuring out whether or not these provisions have been happy New Annex 117 |
A listing of checks, references to analytical procedures, and acceptable acceptance standards which might be numerical limits, ranges, or different standards for the check described. It establishes the set of standards to which a fabric ought to conform to be thought of acceptable for its supposed use. “Conformance to specification” signifies that the fabric, when examined based on the listed analytical procedures, will meet the listed acceptance standards EU Glossary refers a reader to Chapter 4 for the definition57 |
|
Information Threat Evaluation |
The method of evaluating the dangers related to the regulated consumer’s information. It ensures an environment friendly and efficient strategy to information integrity by contemplating the vulnerability of knowledge to involuntary or deliberate alteration leading to risk- based mostly management measures. New Chapter 4 (Line 537-540)7 |
The method of evaluating the dangers related to the regulated consumer’s information. New Chapter 4 (Line 589)7 |
New Annex 11 and Chapter 4 are inconsistent as the previous makes use of ALCOA+ and the latter ALCOA++. USP <1029> replace on good documentation tips and information integrity, issued per week earlier than the New Annex 11 draft, makes use of ALCOA++ with heavy emphasis on traceability.29 Extra element on its traceability is the glue for ALCOA standards will be discovered right here.61
The definition of Industrial off-the-shelf has been copied from OECD GLP 17: … if supplied by a vendor to most people, if obtainable in a number of and an identical copies, and if applied by the check facility administration with or without some customization.26 There are three issues:
- As talked about earlier than, the definition retains the GLP time period of check facility administration.
- Industrial have to be eliminated, as GAMP 5 SE has renamed class 3 software program as customary system elements27 which incorporates open supply software program which isn’t a industrial product.
- Customization implies writing software program code when the proper time period ought to be configuration.
Proper definitions; mistaken place?
There are two definitions (hybrid system and automatic script) within the New Chapter 4 glossary that ought to be included in Annex 11. Why are they in Chapter 4? Automated script can’t be discovered within the physique of Chapter 4 as a result of computerized validation script is used as a substitute. Maybe including validation to the definition is suitable.
The answer?
That is quite simple; all outlined phrases ought to be in a single GMP glossary that’s up to date every time a brand new ultimate chapter or annex is issued.
Abstract
The updates of EU / PIC/S Annex 11 and relevant sections of Chapter 4 signify a significant change and moved from interpretive to prescriptive regulation. That is compounded by the New Annex 11 not aligning with the present model (2011).
We have now a bloated replace with the next potential impacts:
Key sections from the prevailing A11 omitted and many others.
- The applying ought to be validated; IT infrastructure ought to be certified.
- Relegation of The place a computerised system replaces a handbook operation or system to the again of the precept – it ought to be on the entrance!
- No requirement for a system description or stock of methods.
- Automated check instruments and enlargement to incorporate any instruments to help validation.
- Incident administration.
- Glossary issues, inconsistencies and omissions.
Limiting inspection flexibility?
- May an inspection be decreased to a guidelines as a substitute of assessing an organization on a versatile risk-based strategy? This is perhaps the case when inspecting IT safety with the 20 clauses in part 15.
- Will inspectors be skilled to examine all areas lined by the New Annex 11?
Extra importantly, will they’ve technical experience to use throughout an inspection? - PIC/S PI-011 states in 23.4: … inspectors ought to use it selectively to construct up a transparent image of an organization’s scale and complexity of on-site computerization (or automation) and examine selectively the important methods and dangers.36 This flexibility is misplaced with the replace.
- Moreover, it’s important that there’s a case-by-case danger evaluation by way of an inspection.
Excruciating element limits efficient danger administration
- All computerized methods are the identical – apart from the variations.
- It’s the variations that require a versatile and risk-based strategy to preliminary validation and on-going management.
- Nevertheless, the general content material of the revised Annex 11 is way away from an efficient risk-based strategy.
Overlook digital transformation: hybrid methods are OK!
- The Annex 11 idea paper6 talked about digital transformation however the revision comprises no important enlargement on this space.
- Furthermore, there are 19 references to hybrid methods in Chapter 4 (WHY?) and New 13.9 presents a technically illiterate try and hyperlink handwritten signatures on with the underlying e-records.
- An awesome alternative to push Pharma to enhance has been missed.
Affect on regulated customers
- Some corporations that can’t or is not going to interpret a regulation might take the view if it isn’t said – we don’t have to do that. These organizations star in FDA warning letters.
- Some corporations can not interpret laws and say inform us be compliant. Doing nothing isn’t an choice or excuse: discuss to the regulators, learn trade guides, e.g., GAMP 5 second version (SE)27 attend coaching programs or interact a marketing consultant to study and implement compliant computerized methods
One-word summaries of the updates are:
- Prescriptive
- Bloated
- Repetitive
- Nit-picking
- Technically infeasible (OK, two phrases).
The authors seem to have forgotten the KISS precept (preserve it easy, silly). They need to have solely improved or clarified clauses the place vital. E.g. A11.10 has a superb requirement for change management however nothing about configuration administration, which might solely have required a sentence to explain.
Will probably be fascinating to see the ultimate model when issued, as we suspect that EMA / PIC/S should cope with an avalanche of adversarial feedback from all stakeholders.
Acknowledgements
We want to thank, in alphabetical order, Monika Andraos, Markus Dathe, Jim Henderson, Bob Iser, Eberhard Kwiatkowski, Bunpei Matoba, Yves Samson (whereas on vacation!) and Paul Smith for his or her critiques and constructive feedback which have improved our article. Nice work in a really quick timeframe and really a lot appreciated by us!